Disclaimer: This post reflects my personal reading and interpretation of the FDA’s PCCP guidance. It is not affiliated with, endorsed by, or representative of any employer, organization, or institution I am associated with. If you’re making regulatory decisions, read the actual guidance and talk to a regulatory professional.
Imagine you’ve built an LLM-powered system that reads chest X-rays. It flags suspected pneumothorax so radiologists can prioritize urgent cases. The FDA cleared it. Hospitals are using it. And now you’ve collected six months of real-world data that could make the model meaningfully better.
So you retrain. Sensitivity goes up. False positives go down. Patients benefit. But here’s the question that keeps regulatory affairs teams up at night: do you need to go back to the FDA before you ship this update?
Until recently, the answer was almost always yes. Every significant change to a cleared or approved device triggered a new premarket submission. For a technology built on iterative improvement, that’s a brutal bottleneck. The FDA recognized this tension, and their answer is something called a Predetermined Change Control Plan (PCCP).
I spent time working through the FDA’s guidance document on PCCPs, published in its current form in August 2025. The concepts are straightforward once you see them in action, but the document itself is dense. So I’m going to walk through the entire framework using a fictional device I’ll call ThoraxAI, an LLM-enabled chest X-ray triage system. By the end, you should understand what a PCCP is, what goes into one, and how the FDA expects manufacturers to use it.
The problem PCCPs are trying to fix
The FDA regulates LLM-enabled medical devices through the same pathways it uses for any device: Premarket Approval (PMA), 510(k) clearance, or the De Novo classification process. When a manufacturer makes a significant change to a cleared device, they generally need to submit a new 510(k) or PMA supplement before they can market the modified version.
This works fine for hardware. A surgical stapler doesn’t learn from experience. But machine learning models are designed to improve over time, sometimes by retraining on new data, sometimes by adjusting inputs or expanding compatibility. If every retrained model requires a new submission, you get a regulatory queue that can lag months behind the technology.
The FDA’s 2019 discussion paper first floated the idea of a “predetermined change control plan” for Artificial Intelligence and Machine Learning (AI/ML) devices. The concept: let manufacturers specify, up front, exactly what kinds of modifications they plan to make, how they’ll validate those modifications, and what performance criteria must be met. If the FDA reviews and authorizes that plan alongside the original device, the manufacturer can implement those pre-specified changes without filing additional submissions.
Congress codified this concept in the Food and Drug Omnibus Reform Act of 2022 (FDORA), adding Section 515C to the Federal Food, Drug, and Cosmetic Act (FD&C Act). The August 2025 guidance document fills in the details.
Meet ThoraxAI
Let me introduce our fictional device. ThoraxAI is an Artificial Intelligence-Enabled Device Software Function (AI-DSF) that:
- Receives chest radiograph images from hospital Picture Archiving and Communication Systems (PACS)
- Analyzes each image using a convolutional neural network
- Flags suspected pneumothorax cases and moves them to the top of a radiologist’s worklist
- Does not provide a diagnosis; it prioritizes the reading queue
ThoraxAI was cleared through the 510(k) pathway. It was trained and validated on images from five hospital systems, across a demographic distribution representative of the intended use population. Baseline performance: 94% sensitivity, 88% specificity for pneumothorax detection.
The manufacturer, a fictional company called ClearView Medical, wants to keep improving ThoraxAI after clearance. They also want to expand compatibility to additional X-ray machines. Rather than filing a new 510(k) every time they retrain the model or add a compatible device, they include a PCCP in their original 510(k) submission.
What actually goes into a PCCP
A PCCP has three required sections. Each one answers a different question.
| Component | Question It Answers |
|---|---|
| Description of Modifications | What specific changes do you plan to make? |
| Modification Protocol | How will you develop, validate, and deploy those changes? |
| Impact Assessment | What are the risks of these changes, and how will you mitigate them? |
These three pieces are tightly linked. The Description of Modifications tells the FDA what’s changing. The Modification Protocol proves that the manufacturer has rigorous methods to ensure safety after each change. The Impact Assessment ties them together by evaluating the benefit-risk profile of each modification, individually and in combination.

Let me walk through each one using ThoraxAI.
What exactly are you planning to change?
The Description of Modifications needs to be specific. The FDA’s guidance is clear: vague statements like “we may improve performance” won’t cut it. Each modification needs to be defined precisely enough to be verified and validated.
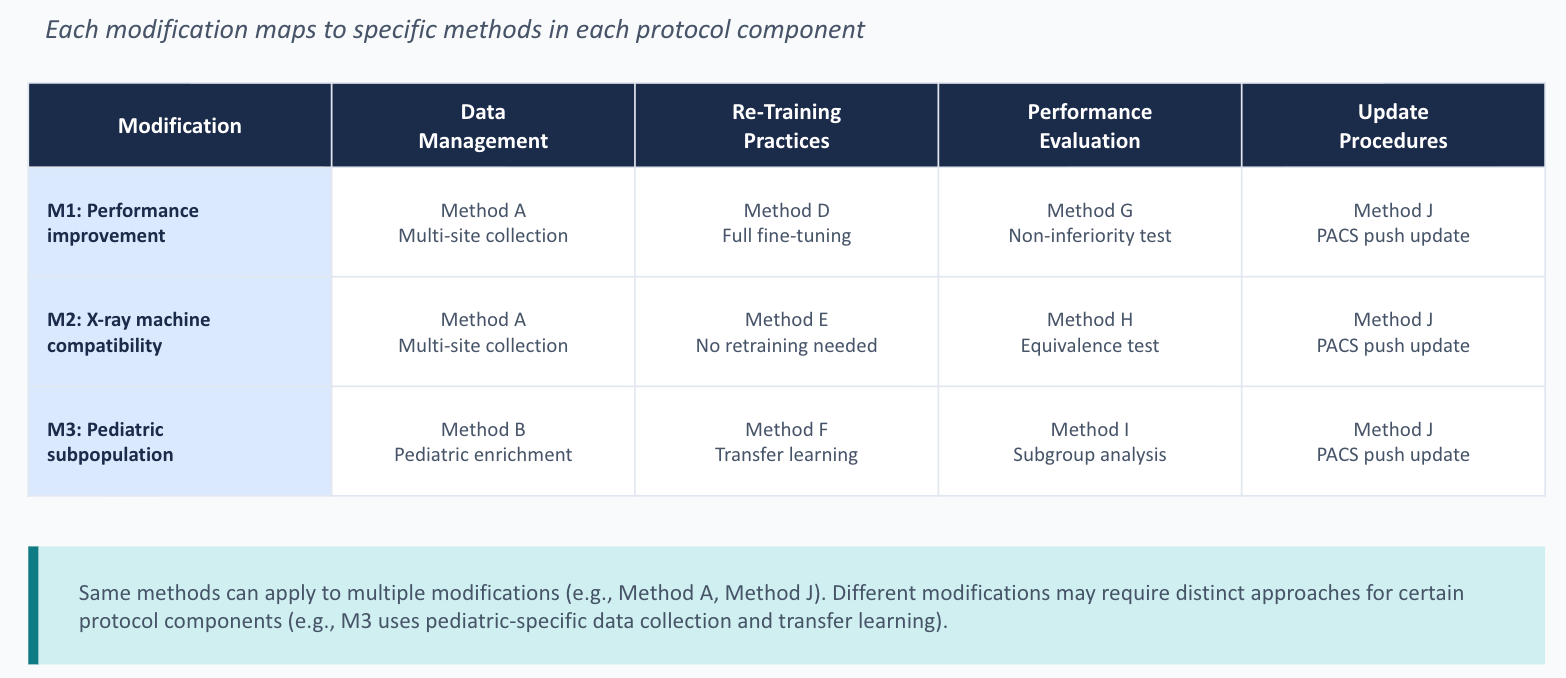
For ThoraxAI, ClearView Medical specifies three modifications in their PCCP:
| Modification | Description |
|---|---|
| M1: Performance improvement through retraining | Retrain the model on new chest radiograph data from additional hospital sites to improve sensitivity and specificity. Sensitivity must remain at or above 94% (non-inferiority margin of 2%). Specificity must remain at or above 88%. |
| M2: Expand X-ray machine compatibility | Extend the AI-DSF to process images from additional X-ray machines that meet defined minimum image acquisition specifications (resolution, bit depth, field of view). |
| M3: Improve performance in pediatric subpopulation | Retrain on a larger pediatric dataset (ages 2 to 17) to improve sensitivity for this subgroup, which was underrepresented in the original training data. Pediatric sensitivity must reach at least 90%. |
Notice a few things about these modifications. They’re specific. They include quantitative acceptance criteria. They don’t change the intended use of the device (it’s still a triage tool for radiologists, not a diagnostic system). And each one can be independently validated.
The Description of Modifications also needs to state whether changes will be implemented automatically (by the software itself, sometimes called “continuous learning”), manually (requiring human action), or some combination. For ThoraxAI, all three modifications are implemented manually: ClearView’s engineers retrain the model, validate it, and push updates to hospitals.
One more detail the FDA wants here: will the modifications be global (the same update goes to every device) or local (different updates for different sites or patient populations)? M1 and M2 are global. M3 is also global, but it specifically addresses a subpopulation. If ClearView wanted to deploy site-specific models, they’d need to describe the local factors that warrant different versions.
How will you actually validate each change?
The Modification Protocol is where the real work lives. It has four components: data management practices, retraining practices, performance evaluation, and update procedures.
Data management
The FDA needs to know how ClearView will collect, annotate, and store the new data used for retraining and testing. Key questions include: Where will the data come from? How will it represent the intended use population across demographics like age, sex, and race? How will the test set be kept separate from the training data?
For ThoraxAI, ClearView’s data management plan might specify that new radiographs will be collected retrospectively from at least three new hospital sites, with consecutive case sampling within defined date ranges. Images will be annotated by a panel of three board-certified radiologists, with majority vote determining the reference standard. Training, tuning, and test sets will be split at the patient level (not the image level) to prevent data leakage. Test data will be sequestered from the development team through an independent data custodian.
The FDA cares deeply about test data independence. If the same people building the model can peek at the test set, even indirectly through iterative testing on the same held-out data, the performance estimates become unreliable. The Modification Protocol needs to describe concrete procedures for sequestration: who has access, what controls prevent unauthorized use, and what happens if the test set needs to be refreshed.
Retraining practices
This section identifies which parts of the model will change and how. For M1, ClearView might describe fine-tuning all layers of the convolutional neural network using the combined original and new training data, with the same architecture and hyperparameters. For M3, the retraining might use a different strategy, such as oversampling pediatric cases or applying transfer learning from the adult model.
The FDA also wants to know what triggers retraining. Does ClearView retrain on a fixed schedule (quarterly, annually)? Or do they wait until a certain volume of new data accumulates? Or is it triggered by observed performance drift? Each approach has different implications for validation, and the Modification Protocol needs to address them.
Performance evaluation
This is the section where you prove the modified device still works. For each modification, the Modification Protocol specifies the test methodology, performance metrics, statistical analysis plan, and acceptance criteria.
For ThoraxAI Modification M1, the performance evaluation might look like this:
| Element | Specification |
|---|---|
| Test dataset | Minimum 1,000 chest radiographs from sites not used in training, with at least 200 positive pneumothorax cases |
| Primary endpoint | Sensitivity for pneumothorax detection |
| Secondary endpoints | Specificity, positive predictive value, area under the ROC curve |
| Acceptance criteria | Non-inferiority of sensitivity (lower bound of 95% CI ≥ 92%) compared to original device; specificity ≥ 88% |
| Statistical test | One-sided non-inferiority test at alpha = 0.025 |
| Subgroup analyses | Performance stratified by age group, sex, and X-ray machine manufacturer |
A critical point the FDA makes: if a modification fails its acceptance criteria and the failure can’t be resolved through root cause analysis, the modification does not get deployed. The Modification Protocol must explicitly state this. You can’t ship a model that didn’t pass its own validation just because the improvement looked promising on the training set.
The performance evaluation also needs to compare the modified device against both the original version (the version cleared by the FDA with no modifications applied) and the most recently modified version. This dual comparison prevents gradual drift where each individual change is small, but the cumulative effect is significant.
Update procedures
How will the updated model actually reach the hospitals using ThoraxAI? This section covers the software deployment process, user communication, labeling updates, and post-market monitoring.
ClearView might specify that software updates are pushed to hospital PACS integration servers during scheduled maintenance windows, with IT administrators confirming installation. Updated labeling, including revised performance claims and a version history, ships with each update. Users receive release notes describing what changed and how performance was affected.
The FDA also expects manufacturers to describe their real-world monitoring plan. How will ClearView track adverse events across different model versions? How will they detect performance degradation in specific subpopulations after deployment? What triggers a rollback to a previous version?
This is where the rubber meets the road for continuous safety. An authorized PCCP doesn’t mean the FDA stops paying attention. It means the manufacturer takes on more responsibility for ongoing validation and monitoring.
What could go wrong, and what’s the plan?
The Impact Assessment ties everything together. It asks: for each planned modification, what are the benefits and risks? How do the Modification Protocol’s validation activities mitigate those risks? And what’s the cumulative impact of implementing all modifications together?
For ThoraxAI, the Impact Assessment might analyze M1 through M3 individually and then consider their interaction. For example, retraining on new data (M1) while simultaneously expanding to new X-ray machines (M2) could introduce confounding effects: if performance drops, is it because of the new training data or the new image sources? The Impact Assessment should describe how ClearView’s validation strategy disentangles these factors, perhaps by validating M1 and M2 independently before combining them.
The guidance also asks manufacturers to consider risks of unintended bias. If the new training data overrepresents certain demographics, the retrained model might improve overall performance while degrading performance for underrepresented groups. The Impact Assessment needs to address how subgroup performance will be monitored and what thresholds trigger corrective action.
Four scenarios that show when the plan works and when it doesn’t
Here’s where the PCCP framework becomes concrete. Let me walk through four post-authorization scenarios for ThoraxAI.
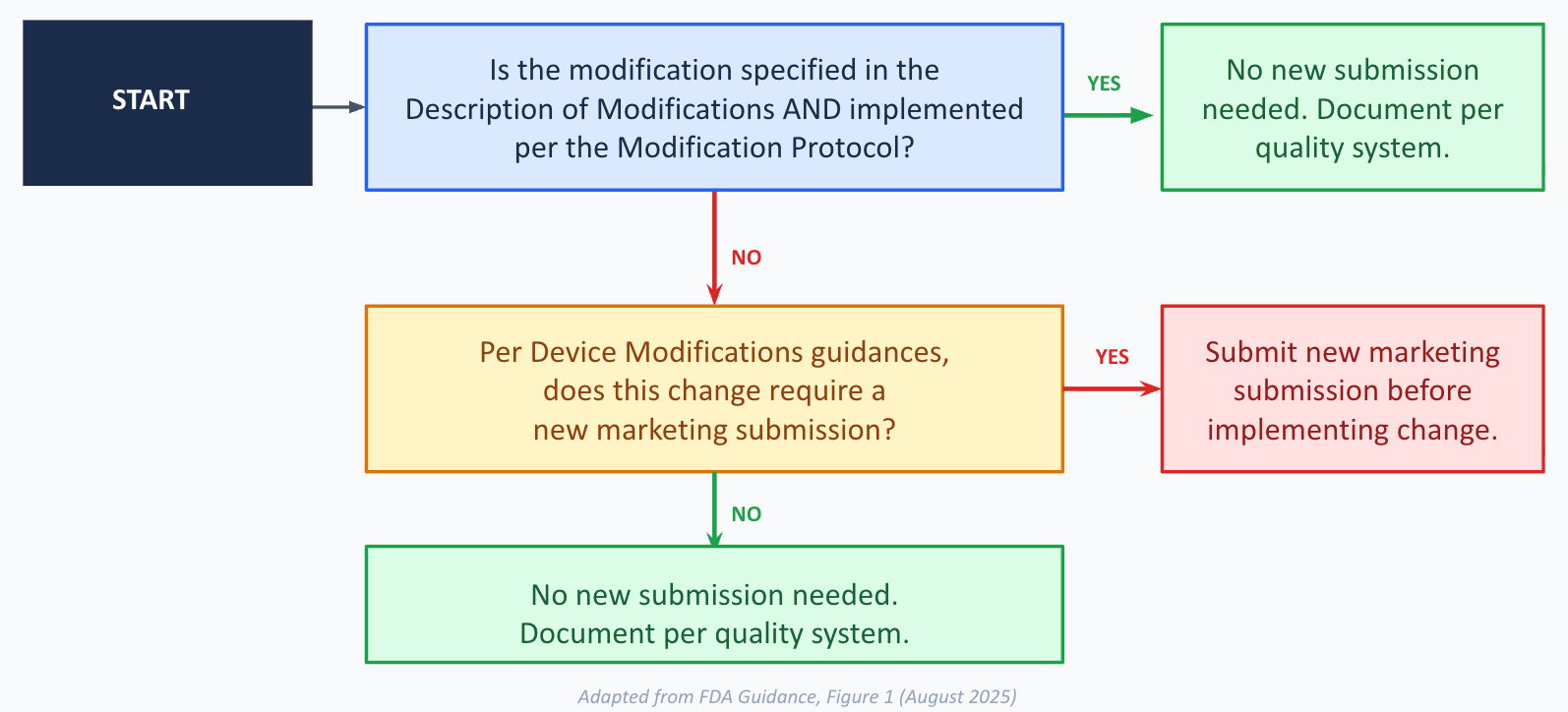
Scenario A: ClearView retrains the model on new data from three additional hospitals. Sensitivity improves to 96%. Specificity holds at 89%. All acceptance criteria in the Modification Protocol are met. Labeling is updated. This is M1, executed exactly as specified. No new 510(k) needed. ClearView documents everything in their quality system and ships the update.
Scenario B: ClearView wants to add compatibility with a new portable X-ray machine that meets the minimum image specs defined in the PCCP. Analytical testing shows equivalent performance. This is M2, executed as specified. No new 510(k) needed.
Scenario C: During retraining, ClearView discovers that the updated model can also detect pleural effusions, which the original model couldn’t. They want to add this as a new output. This was never specified in the PCCP. Detecting a new condition changes the device’s clinical function. A new 510(k) is required.
Scenario D: ClearView follows the Modification Protocol for M1, but the test data was compromised because a team member inadvertently used test images during model tuning. The validation results are unreliable. Even though M1 was specified in the PCCP, the implementation deviated from the Modification Protocol. The modification cannot be deployed under the PCCP. ClearView likely needs a new 510(k) to implement this change, after fixing the data management failure.
The pattern is clear. A modification is “consistent with” the authorized PCCP only when two conditions hold: it was specified in the Description of Modifications, AND it was implemented in conformance with the Modification Protocol. Miss either condition, and you’re back to the standard regulatory path.

The guidance also warns that deviations from an authorized PCCP can render the device adulterated and misbranded under the FD&C Act. This isn’t hypothetical. If a manufacturer ships a model update that doesn’t follow their own PCCP, the FDA can take enforcement action, including seizure or injunction.
What a PCCP can’t do for you
A few important boundaries are worth calling out.
A PCCP cannot change the device’s intended use. ThoraxAI is a triage tool for radiologists. If ClearView wanted to turn it into a diagnostic aid or make it patient-facing, that’s outside the scope of any PCCP. New submission required.
A PCCP sits within the existing regulatory pathway. It doesn’t lower the bar for safety and effectiveness. It just lets manufacturers pre-specify and pre-authorize a defined set of changes so they can move faster when those changes are ready.
Modifications to the indications for use are generally difficult to include in a PCCP. The FDA says most such changes would be hard to assess prospectively. There might be narrow exceptions (like adding compatibility with a specific additional device), but the guidance strongly encourages manufacturers to discuss these through the Q-Submission Program before trying.
And the PCCP itself can change over time, but modifying an authorized PCCP requires its own marketing submission. If ClearView wants to add a fourth modification to their PCCP, say, expanding to CT scans, they’d need to submit a new 510(k) that includes the modified PCCP.
Labeling and telling users what changed
The FDA puts real emphasis on transparency for devices with authorized PCCPs. The labeling needs to tell users that the device incorporates machine learning and has a PCCP, that software updates may modify performance, inputs, or use, and what the current version of the device looks like (including performance data, training data characteristics, and validation results).
As modifications are implemented, the labeling should be updated to reflect what changed, how it was validated, and how users will be informed. Public-facing documents like the 510(k) summary or PMA Summary of Safety and Effectiveness Data (SSED) should also describe the PCCP in sufficient detail for transparency, excluding trade secrets.
This matters because clinicians making decisions based on ThoraxAI’s output need to understand what the device is doing and how it’s evolved. A model retrained six months ago on different data might behave differently in edge cases. The labeling is the primary channel for communicating this.
The quality system angle
One thread that runs through the entire guidance is the manufacturer’s quality system. Under 21 CFR Part 820 (the Quality System Regulation, or QSR), manufacturers must document design changes, maintain device master records, and follow corrective and preventive action processes.
For devices with PCCPs, the quality system isn’t just background compliance. It’s the infrastructure that makes the PCCP work. Every modification implemented under a PCCP must be documented in accordance with the quality system. Design controls, nonconformance handling, version management: all of it applies. The FDA can withhold clearance of a PCCP if there’s evidence that the manufacturer’s quality system can’t support it.
This is a subtle but important point. A PCCP isn’t just a regulatory document. It’s a commitment to a level of process maturity that many smaller companies building LLM-powered medical devices may need to build from scratch.
What I took away from all this
After reading the full guidance, a few things stand out to me.
First, the PCCP framework is a genuine attempt to reconcile the FDA’s safety mandate with the reality of how ML systems improve. The traditional model of “freeze the device, submit a new application for every change” was never going to scale for ML-based devices. The PCCP creates a structured middle ground: the FDA reviews the plan, not every individual update, but the plan has to be detailed and the manufacturer has to follow it precisely.
Second, the emphasis on data representativeness runs deep. The guidance returns to this point repeatedly: training, tuning, and test data must represent the intended use population with respect to race, ethnicity, sex, age, disease severity, and more. Subgroup performance isn’t an afterthought. It’s woven into the data management, performance evaluation, and impact assessment requirements. For anyone working on medical LLM systems, this sends a clear signal about where the FDA’s attention is focused.
Third, the traceability requirements are demanding but sensible. Every modification links to specific validation activities. Every validation activity has predefined acceptance criteria. Every deviation gets documented. The traceability table in the guidance (mapping each modification to its data management, retraining, evaluation, and update methods) is a small thing, but it forces a level of discipline that would improve most ML development workflows even outside the regulatory context.
Finally, the post-market monitoring expectations are real. An authorized PCCP shifts some validation responsibility from pre-market review to the manufacturer’s ongoing operations. That only works if the manufacturer is actively watching for performance drift, adverse events, and emerging biases after each update. The guidance asks manufacturers to describe exactly how they’ll do this, and that’s the right question.

If you’re building medical LLM systems, read this guidance
If you’re building LLM-powered tools for healthcare and you’re not yet thinking about PCCPs, you probably should be. Even if your device is years from a submission, understanding the PCCP framework will shape how you design your ML pipelines, how you manage data, and how you think about version control and validation.
The FDA encourages manufacturers to use the Q-Submission Program to discuss proposed PCCPs before filing. Given that this is still relatively new territory for both industry and the Agency, early engagement seems wise.
The full guidance is available on the FDA’s website. It’s forty-plus pages of regulatory prose, but the Appendices are worth the effort. Appendix A walks through detailed questions for each component of the Modification Protocol, and Appendix B provides six worked examples across different device types: patient monitoring, skin lesion analysis, ventilator settings, image acquisition, feeding tube placement, and a combination product with an imaging drug.
I built ThoraxAI as a teaching example, but the real devices the FDA reviews face all the same questions: what can change, how do you prove it’s still safe, and what happens when something doesn’t go according to plan. The PCCP framework gives manufacturers a way to answer those questions prospectively. The challenge, as with most things in regulated ML, is in the execution.